XRR数据导入拟合
对于XRR的拟合,显然GenX1是更为强大和方便的2。然而也面临着拟合参数选取过于依赖经验,经常过拟合或者无法拟合的现象。 xrayutilities采用lmfit模块进行拟合。我们首先对官方给出的教程进行学习。然后逐步扩展到其他衬底和材料。
数据的导入
1 | import os |
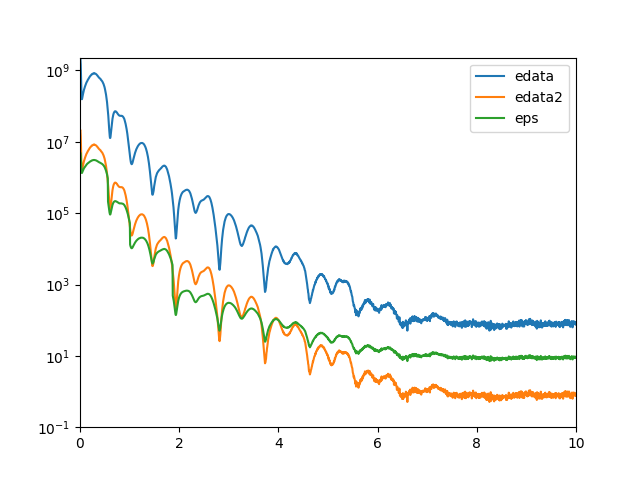
从图中可以看出,强度除以200的值和eps还是有差异的。
定义材料与模型创建
1 | # define layers |
基本没有太多新的知识,只需要根据别的教程建立自己的材料就可以了。至于模型创建中的参数,还需要结合个人具体实验情况。
设定拟合参数
1 | # embed model in fit code |
set_fit_limits应当是 lmfit.Model下的函数,用来设置拟合参数的约束、限制等等。具体参数含义如下:
- name: 参数名称
- vary: 参数在拟合期间是否变化,默认为True
- min、max是模拟的最大最小值,默认无上下限。-numpy.inf,没有下限。numpy.inf,无上限。
拟合并保存拟合结果
1 | # perform the fit |
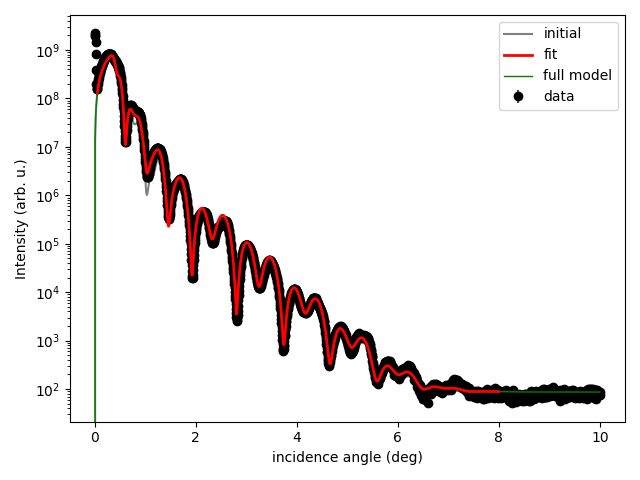
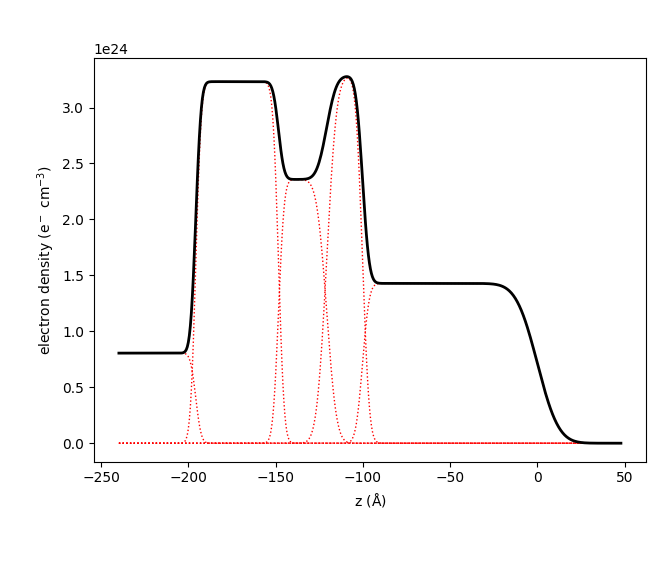
实际应用
建立衬底晶体结构
对\(\ce{YNiO3}\)/\(\ce{YAlO3}\)样品进行拟合,首先是定义材料。
对于\(\ce{YAlO3}\),文献中的晶体结构是Pbnm,然而62空间群常规而言是Pnma。需要将原子坐标\((x, y, z)\)进行转换。也就是给坐标向量乘如下矩阵:
\[\begin{bmatrix} 0 & 0 & 1\\ 1 & 0 & 0\\ 0 & 1 & 0 \end{bmatrix}\]1 | Pbnm空间群的a轴等同于Pnma空间群的c轴; |
因此,材料定义如下: 1
2
3
4
5
6
7YAlO3 = Crystal("YAlO3", SGLattice(62, 5.30102, 7.34181,5.16098,
atoms=[e.Y, e.Al, e.O, e.O],
pos=[('4c', (0.05195, -0.01158)),
'4b',
('8d', (0.2933, 0.0431, 0.7054,) ),
('4c', ( 0.4805, 0.0832 ) )
]))
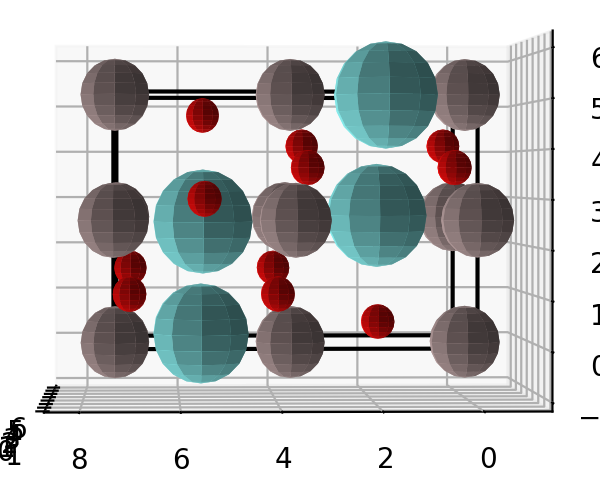
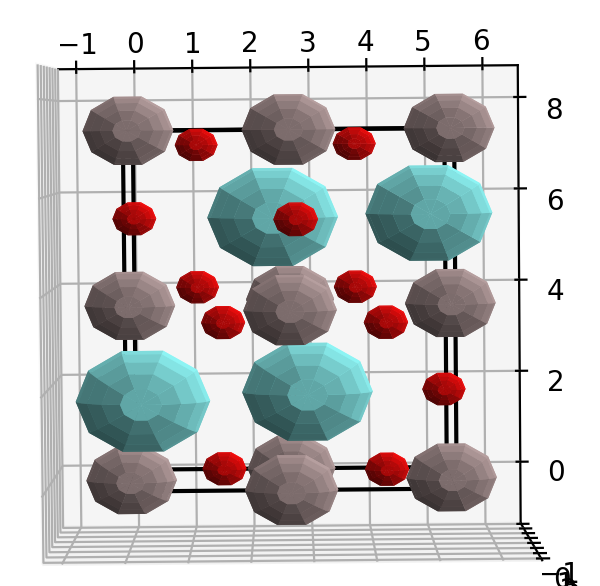
然后,绘制出晶体结构,看构建的晶体是否正确。 1
2
3
4
5
6
7import matplotlib.pyplot as plt
from xrayutilities.materials import predefined_materials
YAlO3 = predefined_materials.YAlO3
f = plt.figure()
YAlO3.show_unitcell(fig=f, subplot=121)

建立薄膜晶体结构
除了自己建立晶体结构以外,还可以导入cif文件。
1 | YNiO3 = xu.materials.Crystal.fromCIF(os.path.join("/data", "YNiO3.cif")) |
然而实际中发现导入cif文件后晶体的命名并非预期情况。因此,可能根据cif文件,自己建立晶体结构是最好的选择。
创建模型
其余与示例一直,注意初始参量对拟合是否收敛同样起到关键性的作用。