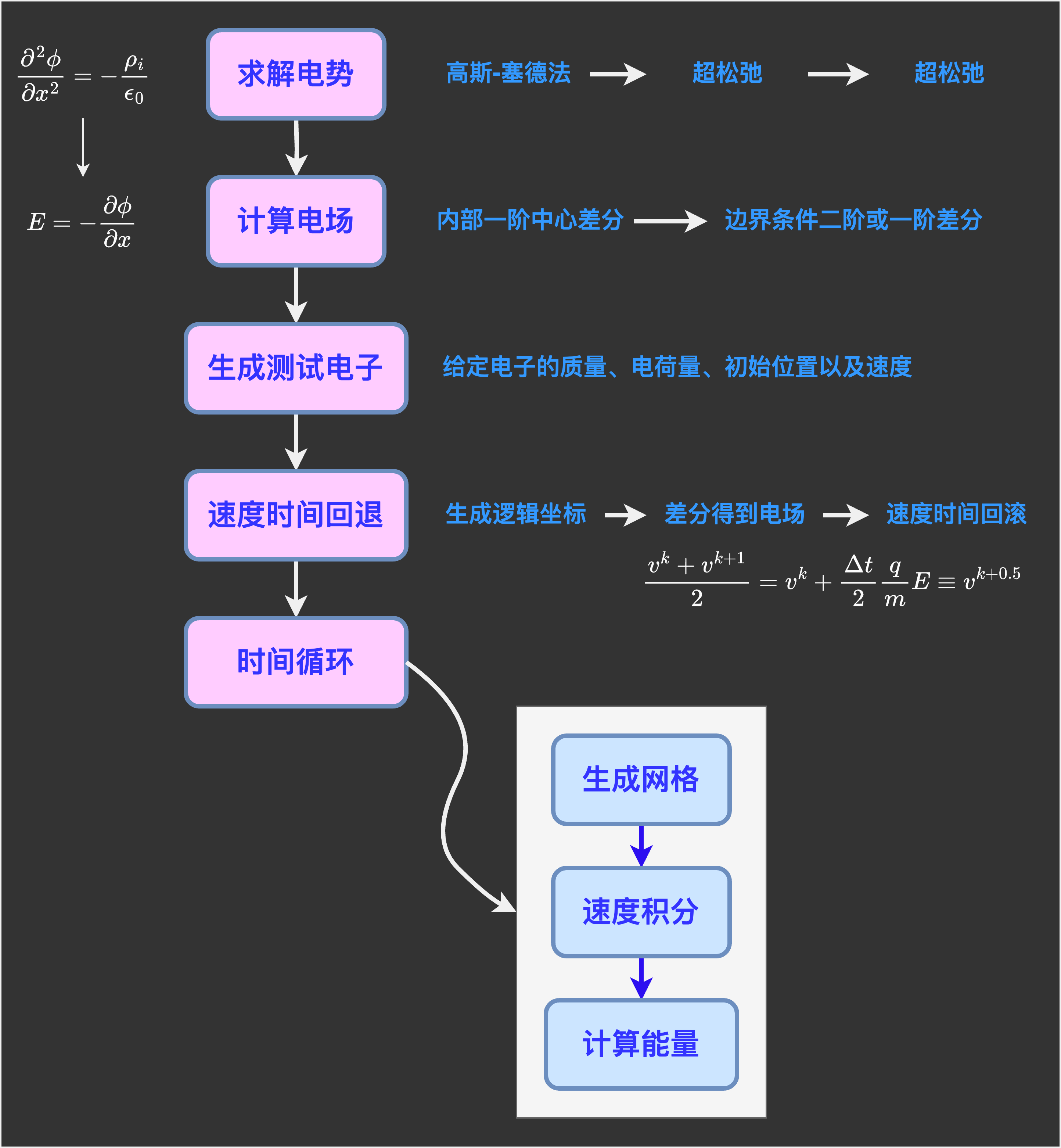
等离子体模拟基础
"Plasma Simulations by Example"一书的学习笔记。
介绍
本章从动力学和流体方法模拟等离子体的概述开始,并介绍了静电PIC控制方程。随后,讨论了有限差分法对微分方程的离散化。我们将使用这种方法来模拟被困在势阱中电子的行为。
气体模拟方法
在微观尺度上,等离子体可以简单地看作由带电粒子(离子和电子)组成的气体。这是我们在本书中采取的基本观点。我们主要关注的是由中性原子、正离子和电子(足以中和空间电荷的)这样的基本组合。 流体模拟的目标是根据一些初始条件和控制定律,预测粒子速度和位置的演变。在经典牛顿物理学的框架下,我们假设所有的中性粒子、离子和电子都作为刚体受运动方程控制,
\[\begin{equation}\label{1.1} \frac{\mathrm{d} \vec{x}}{\mathrm{d} t}=\vec{v} \quad \frac{\mathrm{d} \vec{v}}{\mathrm{d} t}=\vec{a} \end{equation}\]
------Ai 润色到此处
其中,\(\vec{x}\),\(\vec{v}\)和\(\vec{a}\)分别是单个粒子的位置,速度和加速度。如果粒子的质量\(m\)保持不变(本书中该假设均成立),加速度由牛顿第二定律可知为\(\vec{a}=\sum \vec{F} / m\)。 一些力,如重力,来自外部环境。其他力可能是系统固有的力。带电粒子通过库仑力相互作用,
\[\begin{equation}\label{1.2} \vec{F}_{c, i j}=\frac{1}{4 \pi \epsilon_{0}} \frac{q_{i} q_{j}}{\left|\vec{x}_{i}-\vec{x}_{j}\right|^{3}}\left(\vec{x}_{i}-\vec{x}_{j}\right) \end{equation}\] 其中,\(i\)和\(j\)是两个带有\(q_{i}\)和\(q_{j}\)电量的粒子。\(\epsilon_{0} \approx 8.8542 \times 10^{-12}\ \mathrm{C} /(\mathrm{Vm})\) 是自由空间的介电常数。该公式忽略了移动电荷的磁场效应。单个粒子受到的总的力为, \[\begin{equation}\label{1.3} \vec{F}_{c, i}=\sum_{j}^{N} \vec{F}_{c, i j} \quad i \neq j \end{equation}\]
其中,求和是对系统所有粒子的。粒子的速度也可以被碰撞改变。因此,我们有: \[\begin{equation} \frac{d\vec{v}}{dt}=\frac1m\left(\vec{F}_g+\vec{F}_c+\ldots\right)+\left(\frac{d\vec{v}}{dt}\right)_{col} \end{equation}\] 尽管上述公式中包含了重力,但人们习惯上忽略它。
直接方法
从概念上讲,一个包含N个粒子的系统可以通过遍历整个列表来建模,并且每个粒子所受库仑力由下式计算: \[\begin{equation} \vec{F}_{c,i}=q_i\vec{E}_i \end{equation}\] 其中, \[\begin{equation} \begin{array}{rrrrrr}&&\vec{E_i}=\sum_j\frac1{4\pi\epsilon_0}\frac{q_j}{r_{ij}^3}\left(\vec{r_i}-\vec{r_j}\right)&&i\neq j&\\\hdashline\end{array} \end{equation}\]
通过简单的估算可知,这样计算一次的时间比宇宙的年龄还要长七个数量级。实际上,后面会讲到有很快的方法计算。
然而,即使假设线性缩放,由于粒子数量巨大,计算运行时间也将是 21 年。此外,即使一些突然的突破导致计算速度的大幅提高,存储粒子数据所需的内存量也是惊人的。仅存储六个浮点数就需要超过 7000 亿 GB 的 RAM,这些浮点数捕获真空压力下单个立方米中 \(10^{16}\) 个粒子的粒子位置和速度。这超出了任何超级计算机的能力。
玻尔兹曼方程
在一个小的控制体积内瞬间冻结所有的粒子,就可以根据它们的速度分量对它们进行分组。为了完成分组,我们在三维速度空间中定义了一个直方图。每个bin对应于x、y和z的速度分量u、v和w的唯一组合。每个bin的宽度由Δu、Δv和Δw给出。u±0.5Δu、v±0.5Δv和v±0.5Δv的粒子均落入同一bin内。在bin无穷小的极限情况下,离散直方图成为光滑的超空间曲面。该曲面表示速度分布函数,简称VDF。
图1.1给出了仅考虑u和v分量时的一个可能的VDF。函数的值,用表面的高度和阴影表示,表示具有相应u和v速度分量的粒子数。我们清楚地看到大多数粒子都集中在一些平均速度的周围。两条直线u = 0和v = 0将每个速度分量划分为一个负半空间和一个正半空间。v速度以v = 0为中心,因此在y方向上没有净运动。另一方面,x存在明显的漂移。即使存在u < 0的粒子,大部分粒子的u为正。

在没有碰撞的情况下,所有具有相同速度的粒子都具有一致的运动。这是牛顿第二定律的直接结果。这里我们假设控制体积足够小,使得力项不存在空间变化。由于结果对所有群体都是相同的,因此不需要对每个bin中的一个以上的粒子进行速度积分。只有通过碰撞,一部分群体才以独特的方式受到影响。因此,我们写出一个保守项: \[\begin{equation}\label{equ-1-7} \frac{df}{dt}=\left(\frac{\partial f}{\partial t}\right)_{col} \end{equation}\]
这种关系表明,分布函数f的全导数只通过一些尚未定义的碰撞算子发生改变。到目前为止,我们所讨论的分布只对它被采样的小控制体积有效。不同的体积,对应其他空间区域,具有独特的粒子速度组合。分布也随时间演化。因此,VDF是7个变量的函数,\(f = f(x, y, z, u, v, w, t)\)。式中:f为(x, y, z)位置和t时刻速度分量为(u, v, w)的粒子数。该分布可以任意归一化,在这种情况下函数值( f是找到这样一个粒子的概率)。我更喜欢使用非归一化版本,因为它携带了完全描述气体状态所需的所有信息。
利用链式法则改写方程\(\ref{equ-1-7}\)中的导数,得到玻尔兹曼方程: \[\begin{equation} \frac{\partial f}{\partial t}+v\nabla\cdot f+\frac Fm\nabla_v\cdot f=\left(\frac{\partial f}{\partial t}\right)_{col} \end{equation}\]
右端为零的无碰撞方程被称为Vlasov方程。与直接考虑颗粒间相互作用不同,我们可以通过积分这个偏微分方程(PDE)来模拟气体的演化。绝大多数等离子体仿真程序都没有采用这种方法。原因简单且耳熟能详:计算要求仍然大大超出了标准计算系统的能力。这些要求产生于如果保留所有的三个空间和速度分量,则需要存储六维数据。在降维的情况下,直接求解玻尔兹曼方程实际上是非常有吸引力的。我们在第6章考虑这类求解器,即使在保留碰撞的情况下也被混淆地称为Vlasov求解器。
麦克斯韦-玻尔兹曼分布函数
显然,必须有替代方案,因为三维等离子体模拟通常在标准的桌面工作站上进行,网格要细得多。内存需求的大部分来自于每个空间单元中需要的大量速度单元。这些bin中的大部分实际上是空的-这实际上可以在图1.1中看到。因此,可以做一些额外的简化。第一种选择是假设VDF服从一个解析profile。具体来说,我们可以令每个方向上的速度为: \[\begin{equation}\label{equ-1-9} f_M(v)=\frac1{\sqrt{\pi}v_{th}}\exp\left(\frac{-(v-v_d)^2}{v_{th}^2}\right) \end{equation}\]
这就是麦克斯韦-玻尔兹曼分布函数。利用量子理论或统计力学可以推导出它实际上是分子布居数的自然最高熵态。假设你有一个初始充满气体的盒子,里面恰好包含两种类型的粒子:以速度A运动的慢分子和以速度B运动的快分子。如果没有外力作用,并且壁面是完全镜面的,那么速度只能通过碰撞来改变。在动量交换碰撞中,速度较快的分子稍微减速,速度较慢的分子加速。碰撞率也与相对速度成反比。最终,在足够长的时间之后,两个最初离散的粒子群合并为一个没有净动量转移的粒子群。麦克斯韦-玻尔兹曼分布对应于这个末态,到达这个末态的过程称为热化。
利用式\(\ref{equ-1-9}\)的主要好处是,对于每个维度,VDF减少了到只有两个参数:平均(或漂移)速度\(v_d\) 和热速度\(v_{th}\) 。热速度以温度的形式定义,\(v_{t h}=\sqrt{2 k_B T / m}\) 。因此,只需六个浮点数就可以完整地描述每个空间单元中的VDF。通常,我们进一步假设各向同性的温度,将所需的内存减少到仅四个值:漂移速度的三个分量\(\vec{v}_d\),以及单个量级的热速度。这是对初始粗直方图方法所需的8,000个值的巨大改进!但是生活中没有任何东西是自由的,而且这种做法也不是没有重大的弊端。为了使用方程\(\ref{equ-1-9}\),我们要求气体通过一个"足够的时间"到使热化。这一要求并不总能得到满足。碰撞率与数密度成比例,分子在碰撞之间的平均距离由平均自由程给出: \[\begin{equation} \lambda=1/(\sigma n) \end{equation}\] σ为碰撞截面,将在第4章中详细讨论。n为气体数密度。一个重要的因子,称为Knudsen数,将平均自由程与特征长度联系起来: \[\begin{equation} K_n=\frac\lambda L \end{equation}\] 这个长度是“problem specific”。在真空室的模拟中,我们可以令L为真空室的直径。如果\(K_n \ll 1\) ,意味着\(\lambda \ll L\) ,每一个分子在壁与壁之间的运动过程中都会发生许多碰撞。这样的流动被认为是连续的,麦克斯韦VDF可以被认为是有效的。另一方面,如果\(K_n \gg 1\) ,\(\lambda \gg K\),分子之间比彼此更容易与壁面发生碰撞。这种状态描述了自由分子流。碰撞可以完全忽略,气体动力学由壁面效应驱动。最后,如果\(K_n \approx 1\) ,碰撞之间的距离与特征长度相当。在这种情况下,碰撞会发生,但不足以使麦克斯韦形式的VDF具有确定性。这种状态被称为稀薄气体,在等离子体中经常遇到。
流体和动力学方法
因此,我们可以将气体模拟方法分为流体和动力学两大类。流体方法假设VDF具有解析形式的连续体。通过计算分布函数的矩,得到了熟悉的气体特性,如密度或平均速度。这些都是f的积分,随着速度提高到越来越高的幂次,并在整个速度空间上进行计算。 \[\begin{equation} M^k(\vec{x},t)=\int\vec{v}^kf(\vec{x},\vec{v},t)d\vec{v} \end{equation}\]
零阶、一阶和二阶矩导致了质量、动量和能量守恒方程。对于麦克斯韦的VDF,这些矩给出了计算流体力学( CFD )中使用的标准Navier-Stokes方程。
在概率论和统计学中,“moment” 是描述随机变量分布特征的数学概念。一般来说,“moment” 描述了随机变量在不同次幂下的期望值。常见的"moment" 包括:
零阶矩(Zeroth Moment):零阶矩是一个随机变量本身的期望值,即数学期望。对于随机变量 \(X\),其零阶矩 \(E(X)\) 表示 \(X\) 的平均值或期望值。
一阶矩(First Moment):一阶矩是随机变量与一个常数的乘积的期望值。对于随机变量 \(X\),其一阶矩 \(E(X)\) 表示 \(X\) 的加权平均值,其中权重为 X 的概率密度函数。
二阶矩(Second Moment):二阶矩是随机变量的平方的期望值。对于随机变量 \(X\),其二阶矩 \(E(X^2)\) 表示 \(X\) 的平方的平均值。
另一方面,动力学方法不对VDF的形状做任何假设,允许其自洽演化。前面讨论的直接求解器和Vlasov求解器是动力学方法的两个例子。两者均被发现存在计算量过大的问题。幸运的是,存在另一类基于随机(随机)采样的方法。许多速度库中包含零粒子或仅包含少数几个粒子。我们不试图完全求解离散化后的VDF,而是可以生成若干个概率服从分布函数的随机速度样本。该采样过程如图1.2所示。
, a histogram of discrete velocities (gray bars), and random samples (white circles).")
利用解析形状表示一维Maxwell - Boltzmann分布,并与Vlasov求解速度网格和随机采样进行了比较。我们将这些样本视为粒子,并根据运动方程让它们穿过计算域。在这样做的过程中,它们将离散的速度样本移到新的空间区域。每个粒子被分配一个大粒子权重wmp,这样初始的数密度可以用更少的样本来恢复。
大粒子(或特定)权重简单来说就是模拟粒子所代表的真实原子、离子或电子的数量。对于常权重的情形,我们有wmp = Nreal / Nsim。基于该方法的两种流行方案是粒子模拟(Particle in Cell,PIC)方法[15]和直接模拟蒙特卡罗(Direct Simulation Monte Carlo,DSMC)方法[12]。PIC只考虑电磁力引起的加速度。DSMC考虑中性气体,只对碰撞进行建模。这两种方法有许多相似之处,并且经常结合起来模拟部分电离的气体。我们在第4章开展了PIC - DSMC模拟。随机性引入了噪声,可以通过使用更多的样本(粒子)来减少噪声。一旦解停止演化,也可以通过对结果进行长时间间隔的平均来降低噪声。因此,随机方法最适合于达到稳态的问题。
拉格朗日和欧拉描述
PIC和Vlasov求解器都是自洽求解VDF的动力学算法。然而,他们以不同的方式对待气体。当PIC跟随单个粒子在区域内运动时,Vlasov求解器使用一个固定的网格来进化气体。这两个模型被称为拉格朗日模型和欧拉模型。因此,我们可以将气体模拟分为四类:
Fluid / Eulerian:计算流体力学( CFD ),磁流体力学( MHD)
流体/拉格朗日:光滑粒子流体动力学( SPH )
动力学/ Eulerian:Vlasov (直接动力学)求解器
动力学/拉格朗日:粒子模拟( PIC ),直接模拟蒙特卡洛(DSMC)
本文未讨论的SPH方法与PIC方法的相似之处在于用拉格朗日粒子表示气体。但与PIC中每个粒子携带离散速度不同,SPH粒子表示具有麦克斯韦漂移速度和温度的流体包裹体。最后,PIC和DSMC方案使用欧拉空间网格。该网格用于计算粒子间相互作用。最近,使用“dynamic octrees”的无网格粒子方法取得了很大的进展[26]。由于消除了对空间网格的依赖,这种无网格方法使方法真正具有拉格朗日性质。
本书的绝大部分内容都致力于动力学PIC方法的研究。原因有二。首先,流体方法与中性气体方法有许多相似之处。与计算流体力学( CFD )中求解Navier - Stokes方程不同,磁流体动力学( MHD )方程被求解。动力学( CFD )、磁流体动力学( MHD )方程求解。求解方案类似,因为它们都涉及控制质量、动量和能量守恒的偏微分方程组的数值积分系统。主要的区别产生于需要考虑电磁场的演化。许多优秀的书籍已经对相关的集成方法进行了全面的调查。
但为完整起见,第6章对流体求解器进行了说明。流体方法也常用与PIC耦合形成混合方案。在我的电推进领域,直接模拟电子并不常见。取而代之的是“problem-specific complexity”的流体模型。其次,PIC方法的通用性更强。流体方法仅限于连续介质区域。另一方面,PIC可以在整个克努森数范围内使用,尽管它们最适合稀薄和自由分子流。PIC的局限性来自于效率,而不是关于VDF的一些固有假设。将PIC应用于连续介质流动需要比流体方法大得多的计算资源,但最终也得到了相同的结果。反之则不成立,因为流体方法不能模拟速度分布为非麦克斯韦分布的稀薄气体。
流体方法和动力学方法的比较
为了说明流体方法和粒子方法的区别,我们考虑中性气体绕球流动的模拟。从流体方法开始,我们可以利用对气体动力学(或者查阅相应的工具书)的理解,得到关于质量、动量和可能的能量守恒的偏微分方程。计算温度需要能量方程,但通常我们假设温度保持不变。例如,我们可以得到如下密度和动量表达式: \[\begin{equation} \begin{aligned} \frac{\partial n}{\partial t}+\nabla\cdot(n\vec{u})& =0 \\ mn\left[\frac{\partial\vec{v}}{\partial t}+(\vec{v}\cdot\nabla)\vec{v}\right]& =\vec{F}-\nabla p-mn\nu\vec{v} \end{aligned} \end{equation}\]
这些方程的细节目前并不重要。相反,我们应该注意到,两者都包含一个时间导数项∂( ) /∂t和一个或多个仅依赖于空间性质(如\(\vec{F}\)力项或碰撞阻力项\(mnν\vec{v}\) )或其导数(压力梯度项∇p)的项。因此,方程可以被重写成一种形式: \[\begin{equation} \frac{\partial()}{\partial t}=G(X(\vec{x},t)) \end{equation}\]
式中:G为t时刻空间变化的气体状态X (密度、速度、压力等)的函数。从这种形式可以看出,流体模拟涉及密度、速度等微分方程的时间积分。该方案一般涉及将时域划分为由一个小的∆t时间步分隔的状态。将物理域离散为空间网格。这种方法是必要的,因为有限的计算机内存并不能解决连续的现实世界中无限多的信息。
然后进行积分,从初始条件开始,直到满足退出准则。这可能涉及到模拟一个指定的实时,或者模拟达到稳态,其中感兴趣的属性不再变化,∂( ) /∂t = 0。
构成空间域的块被称为cell。cell角被称为节点,在邻居cell之间共享。cell一般可以具有任意形状,但它们不应该重叠。如果单元格的排序遵循逻辑结构,则单元格的集合称为模拟网格或模拟网格。
 and particle (bottom) approach.")
图1.3所示为均匀的笛卡尔网格,该网格为立方、等维单元格,单元格边缘与3个坐标轴对齐。这种类型的网格由于其简单性,在数值模拟中无处不在。
模拟网格使得在感兴趣的属性如密度或速度是已知的地方定义离散点成为可能。最明显的两个选择是节点和cell中心。除非另有说明,本书中开发的所有模拟都使用了以节点为中心的方法。然后将控制方程转换为一种形式,从而令使用基于网格的数据对其进行评估成为可能。
我们还指定了控制气体如何进入和离开计算域的边界条件。例如,在x方向上可以给定恒定的密度\(n = n_0\) 和非零的流速,\(\vec{u}=u_0 \hat{i}\)(在\(x_{min}\) 入口平面上)。我们还指定了嵌入边界的条件。在图1.3中,这些节点通过灰色阴影显示出来。从图中可以推断出,光滑的几何结构变成了退化的“sugarcube” (或楼梯情况)表示。这种无法解析复杂几何是笛卡尔网格的主要局限性。一些解决方法涉及使用自适应网格细化或切割单元格。在第7章中,我们将看到如何利用非均匀网格来解决这个问题。
可以通过在属于球体的节点上设置离子密度为零,\(n = 0\),来模拟离子在表面的中和损失。这是狄利克雷边界条件的一个例子。该条件为未知量规定了实际值。它并非处处适用。
我们事先并不知道下游的密度,\(x = x_{max}\) 面。指定Dirichlet边界条件将是不正确的。相反,我们告诉代码应该充分发展流动,要求在垂直于边界(或法向)的方向上没有变化,\(\partial n / \partial \hat{n} \equiv \partial n / \partial x=0\) 。这是Neumann边界条件的一个例子。其他类型也有,包括Robin和Cauchy,但出现的次数要少得多。
在下半部分可视化的粒子法根本不利用任何宏观控制方程。它只是简单地根据某种源模型将粒子注入到计算域中。这里我们在\(x_{min}\) 面上注入粒子。选择注入粒子的数量,使其达到所需的密度。初始速度由入口速度分布函数采样得到。我们通过离散Δt时间步,积分运动方程1.1来推进粒子的速度和位置。
我们首先通过考虑作用在每个粒子上的力来计算每个粒子的新速度。我们也可以考虑碰撞。然后我们通过积分速度将粒子推向新的位置。边界条件现在控制着单个颗粒的微观行为。中性粒子撞击球体后可能发生漫反射脱落,而离子则可能重新组合成原子。如果模拟的问题具有空间对称性,那么离开计算域的粒子可能被移除或反射回来。
粒子模拟与流体模拟的不同之处在于,在每个时间步,模拟只由大量离散的位置和速度对组成。我们通常感兴趣的是计算体积密度、速度和温度。从微观粒子数据中获得这些宏观性质需要一些额外的平均。这一般是在计算网格的帮助下完成的。由于数密度只是粒子数除以cell体积,因此可以通过计算每个cell中的粒子数得到局部数密度。气速和温度可以通过类似的方式获得。
静电PIC
假设空间某一小区域包含N个真实离子,用局部速度分布函数\(f\)来描述。我们可以通过从VDF中采样\(M\)个模拟粒子来近似这个“population”。这个小得多的样本代表了整个粒子群,因此每个粒子对应于\(w_{mp} = N / M\) 的实际离子或电子。
每个模拟粒子带着离散速度产生,并携带动量\(w_{mp}m\vec{v}\)。同样,对于局域数和电荷密度计算,粒子贡献\(w_{mp}\)个计数和\(w_{mp}q\) 电荷。速度和位置根据质量为\(m\) 、电荷为\(q\) 的单个物理离子或电子的质量和电荷进行更新。为了避免混淆用于移动粒子的电荷和对电荷密度的贡献,将每个大粒子设想为一堆\(w_{mp}\)的真实离子或电子协同移动。
洛伦兹力
粒子按前面讨论的运动方程运动,力按方程1.5计算。然而,我们没有使用库仑定律,而是使用如下方程求解洛伦兹力: \[\begin{equation} \vec{F}=q\left(\vec{E}+\vec{v}\times\vec{B}\right) \end{equation}\]
麦克斯韦方程组
\[\begin{equation} \begin{aligned}\text{Gauss' law:}&\quad\nabla\cdot\vec{E}=\frac\rho{\epsilon_0}\\ \text{Gauss' law for magnetism:}&\quad\nabla\cdot\vec{B}=0\\ \text{Faraday's law:}&\quad\nabla\times\vec{E}=-\frac{\partial\vec{B}}{\partial t}\\ \text{Ampere's law:}&\quad\nabla\times\vec{B}=\mu_0\left(\vec{j}+\epsilon_0\frac{\partial\vec{E}}{\partial t}\right)\end{aligned} \end{equation}\]
此处,\(\rho \equiv \Sigma_s q_s n_s\)是电荷密度,\(\vec{j}\equiv \Sigma_s q_s n_s \vec{v_s}\) 是电流密度。法拉第定律告诉我们,如果\(\partial\vec{B}/\partial t=0\),则\(\nabla\times\vec{E}=0\)。具有零旋度的向量场被称为无旋的。
亥姆霍兹分解也被称为向量微积分的基本定律,进一步告诉我们,任何足够光滑的向量场\(\vec{F}\)都可以分解为无旋(无旋度)和“solenoidal”(无散度)两部分。这两部分可以用一个标量势(对于无旋)和一个矢量势(对于solenoidal势)来定义。 \[\begin{equation} \vec F=-\nabla\phi+\nabla\times\vec A \end{equation}\]
因此,如果磁场恒定(电场无旋),则: \[\begin{equation}\label{1-22} \vec{E}=-\nabla\phi \end{equation}\]
磁场不随时间变化,就是所谓的静电假设。由于安培定律表明磁场由电流产生,因此该条件意味着电流密度足够低,自感应磁场可以忽略不计。此外,如果存在外部磁体,则所施加的磁场不能随时间变化。
泊松方程
静电假设极大地简化了剩余的分析。将方程\(\ref{1-22}\)代入高斯定律可得: \[\begin{equation} \nabla\cdot(-\nabla\phi) =\frac\rho{\epsilon_0} \end{equation}\]
or
\[\begin{equation} \label{1-24} \nabla^2\phi =-\frac\rho{\epsilon_0} \end{equation}\]
这是泊松方程,它构成了cell中静电粒子( ES-PIC )方法的基础。它提供了等离子体电势和电荷密度之间的关系。电荷密度简单地说是单个“species”的数密度按其电荷标度的总和。在只含有单电荷和双电荷离子和电子的系统中,我们有\(\rho= \text{e}( n_i + 2n_{i2}-n_{\text{e}})\),其中e是基本电荷。双电荷离子的密度可以用单电荷布居数(“population”)来替代,\(n_{i2} = kn_{i}\),其中\(k\)为某一分数。
然后我们有:\(\rho=e[(1+2k)n_i-n_e]=e(Z_in_i-n_e)\) ,\(Z_i\)项是平均电离态。
该方法最具吸引力之处在于认识到电荷密度是宏观性质。实际上,这意味着我们依据模拟网格来定义密度。 网格节点的数量G远小于计算粒子的数量M。 运行次数减少至:
计算电荷密度,需要运行M次 (整个粒子循环);
求解泊松方程,需要运行G log(G)次,(整个网格节点的循环);
计算电场,运行G次,(整个网格节点的循环);
粒子位置的积分,M次 (整个粒子循环);
模拟的主循环
对于PIC方法,首先将计算域离散成模拟网格。在每一个时间步,我们使用粒子位置来计算数密度。 然后,使用相应的电荷密度并利用方程\(\ref{1-24}\)来计算等离子体势。 一旦知道电势,就可以从方程\(\ref{1-22}\)中得到电场。 根据电场产生的洛伦兹来更新粒子速度。 新的速度将push粒子到新的位置,然后重复整个过程。 一般的ES-PIC算法遵从如下步骤:
1 | // 初始化 |
模拟主循环会持续运行直至满足某个退出条件。这可能包括完成预设的实际时间,或者达到稳态。循环结束后,我们通常会保存最终结果并进行额外的终态处理。在第七章讨论到的电磁PIC(EM-PIC)遵循类似的流程,但其推进电场和磁场的方法则有所不同
单粒子运动
现在通过简单的例子展示PIC的核心概念。假定两接地电极之间存在电荷密度\(\rho_0\)均匀的粒子,如下图所示。此外,假定电极是无限大的,因此电荷密度是均匀的,且等离子体仅与\(x\)方向有关。这是一个一维问题的例子。

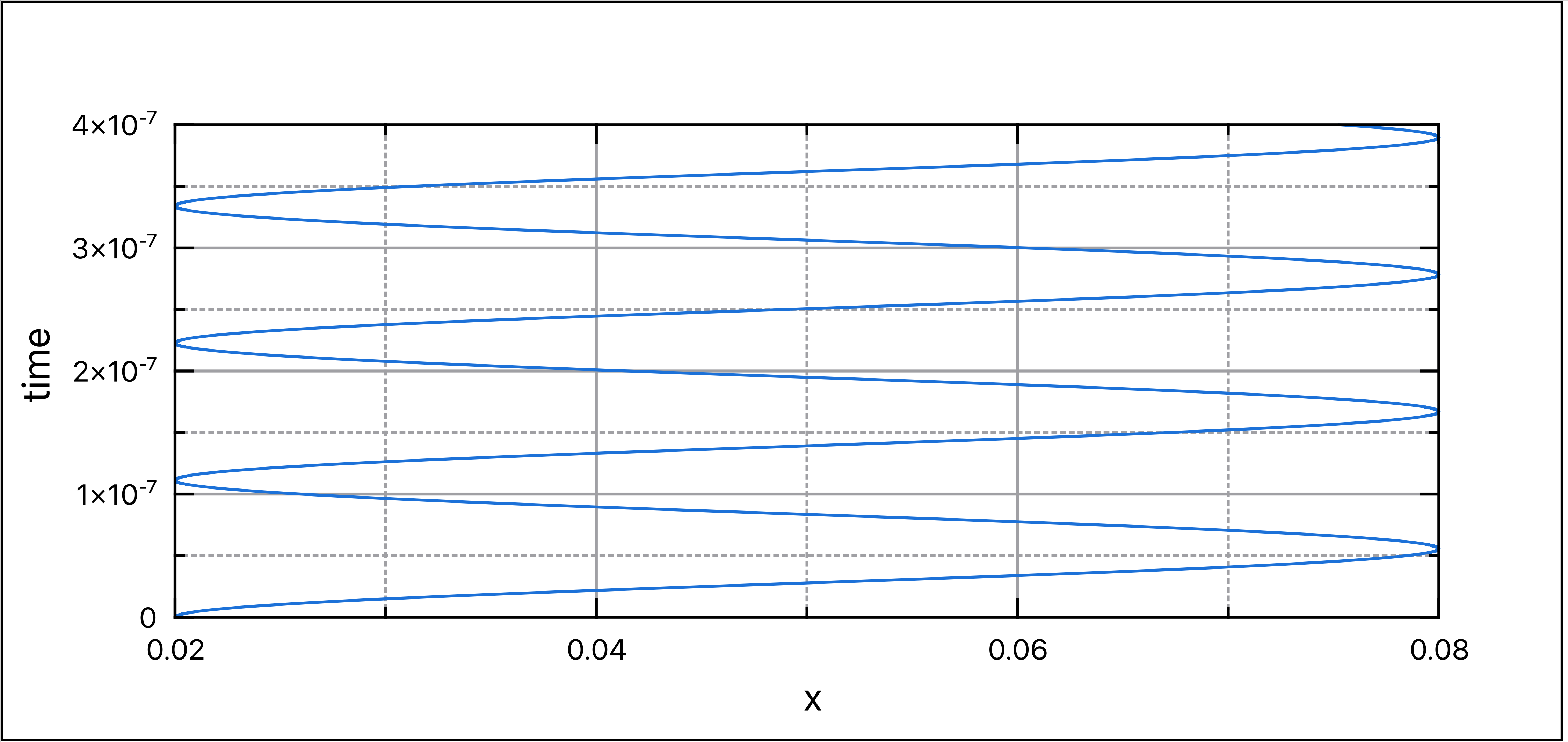
假定该区域仅存在离子,\(\rho_0 = e n_i\)。对于\(\rho_0 >0\),两壁之间的电势为正。 若在靠近某一电极处放一静止电子,那么电子将会向势能顶部移动,在那里达到最大速度。接着它沿山下坡,一路减速,直至速度再次降为零。然后运动方向反转,电子继续以这种来回振动的方式运动。
当然,该模型忽略了任何的摩擦(或碰撞)损失。为了develop模拟,假定背景离子密度保持不变,而且足够高,以至于单个电子所带的电荷对电势分布的影响可以忽略不计。这便使得我们只需计算一次势能。余下的代码仅仅涉及更新电子的位置。我们有如下的伪代码:
1
2
3
4
5
6
7
8求解势
计算电场
载入粒子
主循环:
更新粒子速度
更新粒子位置
write diagnostics
域离散化
如前所述,我们利用计算网格来描述电势、电荷密度以及电场的空间变化。在计算机实现中,这涉及到为每个变量分配一个内存数组,用于存储\(n_i\)个浮点数值,其中\(n_i\)表示网格节点的数量。在经典的C++中,数组可以通过new运算符进行动态分配。
1 | // 常数 |
这种方法有多个缺点。首先,默认情况下分配的数据并未进行初始化。每个数组组件最初仅仅包含分配给它的内存位置上存在的某些随机字节。在使用之前,我们需要手动将每个条目设置为零(或者其他有意义的值)。其次,我们需要记住,在数组不再需要时,使用delete[]运算符来释放内存。
一种更干净的方法是使用C++标准库中的std::vector容器。
1 |
|
你可以注意到两个主要的区别:
不再需要初始化数据。它会被自动设置为零,除非提供了不同的值,如电荷密度那样。
也不需要手动释放内存--当主程序块退出并且向量变量的作用域结束时,这会自动发生。
std::vector
是一个模板容器,能够存储任意类型的数据。
有时候,频繁地输入这些参数可能会变得繁琐。为了解决这个问题,我们可以通过使用指令来定义一个新的类型,叫做dvector。
1 |
|
这种使用关键字的语法是在C++11语言扩展中引入的。另外,我们也可以利用传统的typedef关键字。
1 | typedef dvector vector<double>; // 一个在C++11之前的旧方法 |
至今为止,我们仅仅指定了网格的分辨率。但我们尚未包含与其对应的实际物理区域的信息。 在x方向上均匀分布的笛卡尔网格具有如下位置的节点: \[\begin{equation} x=x_0+i\Delta x\quad i\in[0,c_i] \end{equation}\]
其中\(c_i\)是x方向的cells数量。或者说: \[\begin{equation} \Delta x=L/c_i \end{equation}\]
节点的数量为\(n_i = c_i + 1\),因此上述方程也可以写为: \[\begin{equation} \Delta x=\frac{x_m-x_0}{n_i-1} \end{equation}\]
此处,\(L = x_m -x_0\)表示x值最大的点到原点的距离. 根据图1.4的示意图,我们有\(x_0 = 0\)和$x_m = 0.1 $ m。我们添加代码来设置这些参数。我们还包括一个将网格域输出到逗号分隔文件(.csv)的函数。
1 |
|
编译和运行: 1
2sudo g++ ch1_01.cpp
./a.out
python 代码实现:
1 | from scipy.constants import * |
Julia代码的实现:
1 | using PhysicalConstants.CODATA2018 # 基于国际科学和技术数据委员会(CODATA)发布的2018年常数集 |
有限差分方法
为了得出等离子体势\(\phi\),需要求解泊松方程\(\epsilon_0\nabla^2\phi = -\rho/\epsilon_0\)。 对于此处考虑的恒定电荷密度\(\rho_0 = en_0\)的情况,是有解析解的: \[\begin{equation} \begin{aligned} \frac{\partial^2\phi}{\partial x^2}& =-\frac{\rho_0}{\epsilon_0} \\ \frac{\partial\phi}{\partial x}& =-\frac{\rho_0}{\epsilon_0}x+A \\ \phi & =-\frac\rho{\epsilon_0}\frac{x^2}2+Ax+B \end{aligned} \end{equation}\]
积分常数由边界条件设定。\(x=0\)处\(\phi=0\),因此\(B=0\)。 同样地,\(x=L\)处\(\phi=0\),因此有\(\begin{aligned}A=\rho_0L/(2\epsilon_0)\end{aligned}\)。 则势的解析表达式为: \[\begin{equation} \phi=\frac{\rho_0}{2\epsilon_0}x(L-x) \end{equation}\]
由\(E=-\partial\phi/\partial x\)可得: \[\begin{equation} \begin{aligned}E=\frac\rho{\epsilon_0}\left(x-\frac L2\right)\end{aligned} \end{equation}\]
事实上,实际问题肯定没有如此优雅的解析解。因此,我们必须重写方程从而可以通过计算机进行求解。
有三种主要的可用方法:有限差分方法(FDM)、有限体积法(FVM)以及有限元方法(FEM)。 该书三种方法都有所涉及,先从FDM开始。 对于FDM,我们运用泰勒级数。 \[\begin{equation} \begin{aligned}f(x+\Delta x)&=f(x)+\frac{\Delta x}{1!}\frac{\partial f}{\partial x}+\frac{(\Delta x)^2}{2!}\frac{\partial^2f}{\partial x^2}+\frac{(\Delta x)^3}{3!}\frac{\partial^3f}{\partial x^3}+\ldots\end{aligned} \end{equation}\]
\[\begin{equation} \begin{aligned}f(x-\Delta x)&=f(x)-\frac{\Delta x}{1!}\frac{\partial f}{\partial x}+\frac{(\Delta x)^2}{2!}\frac{\partial^2f}{\partial x^2}-\frac{(\Delta x)^3}{3!}\frac{\partial^3f}{\partial x^3}+\ldots\end{aligned} \end{equation}\]
将上述两方程相加,且令\(f_i\equiv f(x_i)\)、\(f_{i+1} \equiv f(x+\Delta x)\)以及\(f_{i-1}\equiv f(x-\Delta x)\),则有: \[\begin{equation} \begin{aligned}f_{i+1}+f_{i-1}=2f_i+\Delta^2x\frac{\partial^2f}{\partial x^2}+\text{HOT}\end{aligned} \end{equation}\]
HOT是高阶项(Higher Order Terms),此外\(\Delta^2x \equiv(\Delta x)^2\)。由于\(\Delta x<<1\),因此上述方程等号右侧第二项可忽略。 从而得到的方程是二阶精确的。我们使用一个"大O "符号来识别这个截断误差,记为\(O(2)\)。
下一步可以重新书写上述方程以分离导数项。 \[\begin{equation}\label{1-36} \begin{aligned}\frac{\partial^2f}{\partial x^2}=\frac{f_{i-1}-2f_i+f_{i+1}}{\Delta^2x}+O(3)\end{aligned} \end{equation}\] 这个表达式被称为二阶导数的标准中心差分。
势求解器
泊松方程在域内处处成立。在每个内部节点上我们都有 \[\begin{equation} \begin{aligned}\frac{\partial^2\phi}{\partial x^2}=-\frac{\rho_i}{\epsilon_0}\quad i\in[1,n_i-2]\end{aligned} \end{equation}\]
根据方程\(\ref{1-36}\)可将上式改写为: \[\begin{equation} \frac{\phi_{i-1}-2\phi_i+\phi_{i+1}}{\Delta^2x}=-(\rho_i/\epsilon_0)\quad i\in[1,n_i-2] \end{equation}\]
对于\(n_i\)个未知量有\(n_i-2\)个方程。通过指定最左端和最右端节点的边界条件来close the system。 由于两端节点上的电势是固定的,因此采用狄利克雷边界,\(\phi_0=\phi_{ni-1}=0\text{ V}\)。 作为一个例子,我们考虑一个只有七个节点的网格。泊松方程的有限差分法公式产生了下面的方程: \[\begin{equation} \begin{aligned} \phi_0& =\phi_{\boldsymbol{left}} \\ (1/\Delta^2x)\left(\phi_0-2\phi_1+\phi_2\right)& =-\rho_1/\epsilon_1 \\ (1/\Delta^2x)\left(\phi_1-2\phi_2+\phi_3\right)& =-\rho_2/\epsilon_2 \\ (1/\Delta^2x)\left(\phi_2-2\phi_3+\phi_4\right)& =-\rho_3/\epsilon_3 \\ (1/\Delta^2x)\left(\phi_3-2\phi_4+\phi_5\right)& =-\rho_4/\epsilon_4 \\ (1/\Delta^2x)\left(\phi_4-2\phi_5+\phi_6\right)& =-\rho_5/\epsilon_5 \\ \phi_{6}& =\phi right \end{aligned} \end{equation}\]
我们可以用矩阵表示法重写这个system (入下图所示)。

任何线性的矩阵系统可以通过在两边乘矩阵的逆进行求解,\(\mathbf{A}^{-1}\mathbf{A}\vec{\phi}=\mathbf{A}^{-1}\vec{b}\)。 这个公式假设逆矩阵存在:矩阵不奇异。矩阵的逆与矩阵相乘就是单位矩阵,因此有\(\vec{\phi}=\mathbf{A}^{-1}\vec{b}\)。 然而,求解矩阵的逆计算量非常的大。此外,此外,泊松方程的系数矩阵仅由以主对角线为中心的少数非零带组成。 这是带状矩阵的一个例子,它本身就是稀疏矩阵的一种类型。 通过只记录非零元素,稀疏矩阵可以被有效地存储。逆矩阵失去了这种带状结构,并且包含了更多的非零元素。
Tridiagonal 算法
大多数代码中采用的方法是计算给定系统的解,而不需要显式地计算逆。矩阵求解器可以分为两类:直接求解器和迭代求解器。 直接求解器利用矩阵数据中的一些固有结构,在固定的迭代次数内产生精确解。 我们令\(\vec{a}\)和\(\vec{c}\)分别代表主对角线\(\vec{b}\)左边和右边的系数,并标记右边的向量为\(\vec{d}\)。 算法由两步组成,首先,向前sweep,我们将系数修改为:
\[\begin{equation} \left.c_i'=\left\{\begin{array}{ll}\frac{c_i}{b_i},&;i=0\\\frac{c_i}{b_i-a_ic_{i-1}^{\prime}}&;i=1,\ldots,n-2\end{array}\right.\right. \end{equation}\] 以及 \[\begin{equation} \left.d_i^{\prime}=\left\{\begin{array}{ll}\frac{d_i}{b_i},&;i=0\\\frac{d_i-a_id_{i-1}^{\prime}}{b_i-a_ic_{i-1}^{\prime}}&;i=1,\ldots,n-2\end{array}\right.\right. \end{equation}\]
然后,向后代换得: \[\begin{equation} \begin{aligned}x_{n-1}&=d_{n-1}^{\prime}\\ x_i&=d_i^{\prime}-c_i^{\prime}x_{i+1}\ \ \ \ \ \ ;i=n-2,\ldots,0 \end{aligned} \end{equation}\]
算法实现:
1 | void solvePotentialDirect(double dx, dvector &phi, const dvector &rho){ //void 函数返回类型关键字,表示函数不会返回任何值 |
Jacobi 迭代
对于2D和3D问题,经常使用迭代求解器来求解。对于Jacboi迭代,迭代形式为(根据Jacboi迭代可轻松得出,此处不展开): \[\begin{equation} \phi_i^{k+1}=\left[b_i-\sum_{j=0}^{nj-1}(1-\delta_{ij})a_{ij}\phi_j^k\right]/a_{ii} \end{equation}\]
where 对于\(\delta_{ij}\),当\(i=j\)时为0,反之为1。\(k\)和\(k+1\)分别代表当前和新的值。 在这里,利用系数矩阵的稀疏结构也是很有意义的。 此外,对于泊松方程,所有非边界系数都是相同的,因此有: \[\begin{equation} \label{equ-1-51} \phi_i^{k+1}=\left(\phi_{i-1}^k+\phi_{i+1}^k-b\Delta^2x\right)/2 \end{equation}\]
Jacobi 迭代算法实现如下: 1
2
3
4
5
6
7
8
9
10
11
12
13
14for (int k=0; k<max_it; k++){
// 在整数节点上计算k+1的值
for (int i=1; i<ni-1; i++){
phi_new[i] = (phi[i-1] + phi[i+1] - (dx*dx)*b[i])/2;
}
// 应用边界条件
phi_new[0] = phi_left;
phi_new[ni-1] = phi_right;
// 将新的k+1值copy down到 'k'
for (int i = 0; i<ni; i++){
phi[i] = phi_new[i]
}
}
Gauss-Seidel 方案
Jacobi迭代需要存储当前和新的值,然后将新的值覆盖当前值。但我们是否可以直接覆盖当前值?这将给我们Gauss-Seidel方案。 方程\(\ref{equ-1-51}\)被如下方程替代: \[\begin{equation} \phi_i^{k+1}=\left(\phi_{i-1}^{k+1}+\phi_{i+1}^k-b\Delta^2x\right)/2 \end{equation}\]
相应的伪代码: 1
2
3
4
5
6
7
8
9
10// 应用边界条件
phi[0] = phi_left;
phi[ni-1] = phi_right;
for (int k=0; k<max_it;k++){
// 在整数节点计算'k+1'的值
for (int i=1; i<ni-1;i++){
phi[i] = (phi[i−1] + phi[i+1] − (dx*dx)*b[i])/2;
}
}
对角优势:每个方程中对角线系数的绝对值必须大于方程中其他系数的绝对值之和。
当方程组具有对角优势的时候,肯定收敛。如果不具有,也有可能收敛(充分不必要条件)。
连续超松弛
我们可以通过实施连续超松弛( Successive Over Relaxation,SOR )来进一步加快收敛速度。 该方案利用旧的和新的近似来预测未来的解。对于任意两点y1和y2,我们可以用线性拟合定义另一点,\(y = y_1 + w(y_2-y1)\)。 式中:\(w\)为松弛因子。该表达式在\(w = 0\)时取值为\(y_1\),在\(w = 1\)时取值为\(y_2\)。当\(w > 1\)时,我们得到了超出\(y_2\)极限的插值值。 通过这样的方法将新的值和旧的值进行加权平均,也许在\(t>1\)时可以得到比较好的近似。 SOR加速的GS算法为:
1 | for (int k=0; k<max_it; k++){ |
最优加速度参数w的值需要通过试凑来确定,但习惯上使用1.4左右的值。过大的w会导致初始误差向错误的方向传播而出现收敛问题。利用w < 1被称为欠松弛,并在CFD中用于稳定可能发散的求解器。一种自适应的方法有时是首选的。
超松弛隐含了一个假设:新值正在朝着真正解的正确方向移动。
收敛性检查
在迭代次数\(k\)时当前的矩阵系统为: \[\begin{equation} \mathbf{A}\vec{\phi}^k=\vec{b}^k+\vec{R}^k \end{equation}\]
\(\vec{R}^k\)是残差。
通过向量范数来刻画它的大小。有几种范数,其中一个称为L2: \[\begin{equation} ||R||=\sqrt{\frac{\sum_i^n(R_i)^2}n} \end{equation}\]
由于这种计算需要耗费一些宝贵的计算资源,我们一般不会在每次迭代中计算残差。我一般每隔25~50次求解器迭代就对Gauss-Seidel求解器中的残差进行评估,因为这似乎是在不太频繁地执行收敛检查与超出期望的容差运行额外的迭代之间的一个很好的妥协。完整的求解器代码如下所示。至多运行max_it次迭代。一旦范数下降到10-6以下,函数以成功代码终止。否则,如果超过求解器的最大迭代次数而没有达到期望的容忍度,我们返回错误来表示失败。
1 | bool solvePotentialGS(double dx, dvector &phi, const dvector &rho, int max_it=5000){ |
电场
下一个步骤计算电场: \[\begin{equation} E=-\frac{\partial\phi}{\partial x} \end{equation}\]
在节点位置\(i\)处使用泰勒展开:
\[\begin{equation}\label{1-56} \begin{gathered} \boldsymbol{f_{i+1}} =f_i+\frac{\Delta x}1\frac{\partial f}{\partial x}+\frac{(\Delta x)^2}2\frac{\partial^2f}{\partial x^2}+\text{HOT} \\ \boldsymbol{f_{i-1}} =f_i-\frac{\Delta x}1\frac{\partial f}{\partial x}+\frac{(\Delta x)^2}2\frac{\partial^2f}{\partial x^2}+\text{HOT} \end{gathered} \end{equation}\]
\[\begin{equation} \begin{aligned}f_{i+1}-f_{i-1}=2\Delta x\frac{\partial f}{\partial x}+\text{HOT}\end{aligned} \end{equation}\]
同样地,通过一阶偏微分方程的中心差分,可得: \[\begin{equation} \frac{\partial f}{\partial x}\thickapprox\frac{f_{i+1}-f_{i-1}}{2\Delta x} \end{equation}\]
如果忽略二阶导数,则得到向前以及向后差分: \[\begin{equation} \begin{aligned}\frac{\partial f}{\partial x}&\approx\frac{f_{i+1}-f_i}{\Delta x}\\\\\frac{\partial f}{\partial x}&\approx\frac{f_i-f_{i-1}}{\Delta x}\end{aligned} \end{equation}\]
显然: \[\begin{equation} \begin{aligned}f_{i+2}=f_i+2\Delta x\frac{\partial f}{\partial x}+2\Delta^2x\frac{\partial^2f}{\partial x}\end{aligned} \end{equation}\]
对方程\(\ref{1-56}\)乘以4,则得: \[\begin{equation} \begin{aligned}\frac{\partial f}{\partial x}&\approx\frac{-3f_i+4f_{i+1}-f_{i+2}}{2\Delta x}\end{aligned} \end{equation}\]
\[\begin{equation} \begin{aligned}\frac{\partial f}{\partial x}&\approx\frac{f_{i-2}-4f_{i-1}+3f_i}{2\Delta x}\end{aligned} \end{equation}\]
1 | // 通过差分电势计算电场 |
粒子运动
既然我们已经计算出了电势和电场,我们就可以引入试验粒子。我们通过质量、电荷、位置以及速度来定义粒子。 粒子运动由运动方程\(\ref{1.1}\)给出,力由\(F_x=qE_x\)确定。注意,此处不含磁场。 我们使用有限差分,注意时间只能向前流动,因此不能使用中心差分。 \[\begin{equation} \begin{aligned}&x^{k+1}=x^k+v^k\Delta t\\&v^{k+1}=v^k+(q/m)E^k\Delta t\end{aligned} \end{equation}\] 这个方案被称为前向欧拉方法。这是一种显式方法,因为在时间\(k+1\)的新值仅依赖于时间k的数据。 因此,可以为每个粒子定义一个方程式,明确提供新的位置。 这与隐式方法形成对比,在隐式方法中,\(k+1\)处的值依赖于其他也是\(k+1\)处的值。 通常,隐式方案会导致线性系统出现,需要矩阵求解。 在上述公式中,\(E_k = E(x^k)\)表示在第k时间步粒子位置的电场采样。 我们很快会了解如何将网格数据插值到粒子。现在,假设电场是恒定的。通过以下代码展示积分过程:
1 | int main(){ |
向前/向后欧拉方法
原书中讨论了这两种方法,总的来说都有较大的误差。
蛙跳方法
在积分过程中,\(v^k\)和\(v^{k+1}\)是可用的,我们可以利用它们的平均值。 因此我们有: \[\begin{equation} x^{k+1}=x^k+\left(\frac{v^k+v^{k+1}}2\right)\Delta t \end{equation}\] 该方案可以产生更精确的结果。利用\(v^{k+1} = v^{k} + \Delta t(q/m)E\),我们有: \[\begin{equation} \frac{v^k+v^{k+1}}2=v^k+\frac{\Delta t}2\frac qmE\equiv v^{k+0.5} \end{equation}\]
位置需要在半个步时来进行速度积分。由于这些半步时刻仅需要速度,因此让初始粒子速度在\(k=-0.5\)时定义是合理的。
这种方法避免了显式执行平均的需要。虽然这看起来可能是个微不足道的优化,考虑到在典型的PIC模拟中有数百万的粒子以及数十万的时间步长,消除任何低效对我们都是有利的。 通过这种方案,我们知道了在时间k = −0.5、0.5、1.5等时刻的速度。同时,我们也知道了在时间k = 0、1、2等时刻的粒子位置。
1 | int main(){ |
由于回退时间步,实际上是让电场回退到两个格点中间,因此需要差值计算。
替代方法
另一种替代方法是四阶精确的龙格-库塔方法(RK4)。对于一个一般的偏微分方程来说: \[\begin{equation} \begin{aligned}\frac{\partial f}{\partial t}=g(t,f)\end{aligned} \end{equation}\]
未来时间步的值由下式确定: \[\begin{equation} \begin{aligned}f^{k+1}&=f^k+\frac16\left(k_1+2k_2+2k_3+k_4\right)\end{aligned} \end{equation}\] 其中, \[\begin{equation} \begin{aligned} &\boldsymbol{k_1} \Large=\Delta tg\left(t^k,y^k\right) \\ &\boldsymbol{k}_{2} =\Delta tg\left(t^k+\frac{\Delta t}2,y^k+\frac{k_1}2\right) \\ &{\boldsymbol{k}_3} =\Delta tg\left(t^k+\frac{\Delta t}2,y^k+\frac{k_2}2\right) \\ &{\boldsymbol{k}_4} =\Delta tg\left(t^k+\Delta t,y^k+k_3\right) \end{aligned} \end{equation}\]
但是对于等离子体模拟而言,PK4是不常用的,因为它的计算成本很高。具体分析见原书。
插值
上文,我们在格点中计算电场。但对处于格点中间的粒子而言,如果直接使用会导致电场非常不连续。 这种不连续性可能会引发数值不稳定性,导致非物理结果。 作者采用了线性插值的方式,使得电场变得连续。
\[\begin{equation} \begin{aligned}f=f_i+d_i(f_{i+1}-f_i)\end{aligned} \end{equation}\] 上式中,\(f_i\)是格点\(i\)上的电场值,\(f_{i+1}\)是格点\(i+1\)上的电场值,\(d_i\)是插值系数。
可以重新写为: \[\begin{equation}\label{1.79} \begin{aligned}f=(1-d_i)f_i+(d_i)f_{i+1}\end{aligned} \end{equation}\]

上述表达式给出的操作被称为gather,因为它将多个基于网格的值聚集到特定位置以evaluate一个量。 相反的过程被称为scatter,它用于将基于粒子的数据存放回网格。我们在下一章中使用scatter操作来计算粒子密度。 在方程\(\ref{1.79}\)中,\((1−di)\)和\((di)\)项被称为基函数,或者称为形状因子。它们在图1.10中以两条虚线表示。在粒子位置\(x_p\)处,它们的幅值用于缩放两个周围节点\(f_i\)和\(f_{i+1}\)的值。
聚集和分散算法都需要知道包含粒子的单元。我们还需要知道粒子在单元内的相对位置。
对于均匀网格,这种计算很简单。通过反转节点位置的映射,我们得到\(l_i = (x−x_0)/\Delta x\)。这里的\(l_i\)是逻辑坐标。
它只是我们之前用于计算节点位置的整数索引\(i\)的浮点数对应物。 \(l_i\)的整数部分是节点索引,而小数部分是与下一个节点的分数距离\(d_i\)。因此,我们定义一个辅助函数:
1
2
3double XtoL(double x, double dx, double x0=0){
return (x-x0)/dx;
}1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19double gather(double li, const dvector &field){
int i = int(li);
double di = li-i;
return field[i]*(1-di)+field[i+1]*di;
}
我们使用如下函数:
```cpp
dvector ef(ni); // 基于节点的电场
for (int ts=0; ts<2000; ts++){ // 2000个时间步长
// 粒子位置的电场
double li = XtoL(x, dx);
double ef_p = gather(li, ef);
// 速度和位置积分
v += (q/m)*ef_p*dt;
x += v*dt;
}
诊断
我们现在已经实现了完成代码所需的所有部分。但是,就像任何计算机模拟一样,验证结果是否具有物理意义是很重要的。
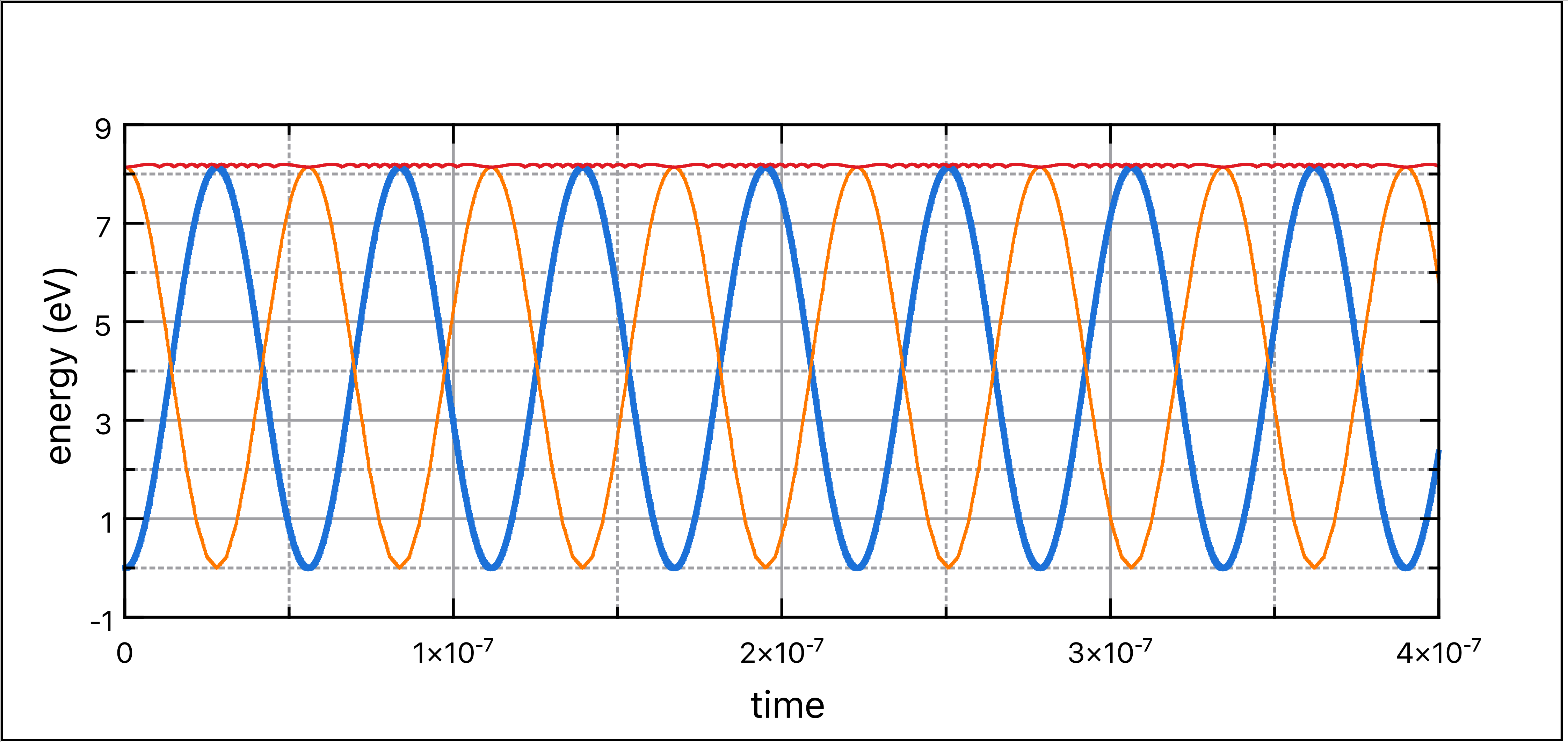
粒子振荡源于能量的守恒。因此,我们应该检查能量是否确实得到了守恒。
在时刻\(k\),一个粒子的总能量由其势能和动能之和给出:
\[\begin{equation}
\frac12mv^2+q(\phi-\phi_{max})=C
\end{equation}\] 其中\(C\)是常数。实现这个诊断检查要求在同一物理时间评估速度与位置。
通过我们的leapfrog,这两个参数实际上有半个时间步长的offset,速度落后于位置。
一种可能性是通过追踪旧位置来使两个参数处于相同的物理时间,在\(x^{k-0.5}=(x^k+x^{k-1})/2\)。
将两种能量从焦耳转换为电子伏特,方法是将它们除以基本电荷。
通过一下代码实现: 1
2
3double phi_p = gather(XtoL((x+x_old)/2, dx) phi); // phi(x(k-0.5))
double ke = 0.5*m*v*v/QE; // KE in eV
double pe = q*(phi_p-phi_max)/QE; // PE in eV
全部代码
1 |
|
python实现(numba加速)
1 | from scipy.constants import * |
Julia实现
1 | using LinearAlgebra |
小结
等离子体计算的流程图